Or, Sometimes Sequels Really are Better
One source of flak ML often gets goes something like this: “Ok, ok, these magical chatbots and robo-Picassos are cool and all, but how can ML actually help the world?”
Fair enough.
One area in desperate need of a shakeup is drug discovery. This field is notoriously slow, often taking decades, all while racking up millions in research costs, for a trial drug that, statistically, won’t pan out in the end. The stakes hardly need introduction – a cure for cancer, faster vaccine development, next-gen heart medications. One of, if not the major bottleneck in drug discovery is protein structure prediction. Protein structure prediction means going from a 2D sequence to the 3D protein shape.
Knowing the 3D structure helps to build more effective medicines and vaccines that can interact with that protein or molecule. As you may remember from biology class, this is like knowing the shape of a lock, so you can build a key to match it.*
* Though this analogy isn’t 100% accurate because enzymes can undergo conformational changes upon binding, but hey, it’s a useful visualization all the same.

Way too many locks means we need a better way of finding the keys. And, unlike Parisian “Love Bridges,” you can’t just chainsaw your way to a solution.
Predicting Protein Structure Is Very Hard
You might ask, shouldn’t similar proteins be basically the same from 2D -> 3D? Alas, no. There are a lot of physical interactions at tiny scale, so a small sequence change can have a really big effect. Proteins with overall similar 2D sequences can look wildly different in 3D form, and proteins with similar shapes can have very different 2D sequences.
Next, you might ask, can’t you just take a picture? In fact, yes! X-ray crystallography has been around for a century. It requires you to crystallize many copies of a protein, shine some x-rays on the crystals, look at the pattern from the refracted light, and infer the protein structure. A more recent version of this process, cryo-electron microscopy, does something similar with fewer steps. In any case, these technologies are very expensive (microscopes in the millions $$!), are time-consuming, and don’t work for very flexible proteins. When we think about drug discovery, it’s a needle in a haystack. The current protein database has around 180,000 proteins. We humans make an estimated 6 million isoforms of proteins from our DNA. Oof.

This bad boy will set you back a cool $8 million.
Ok, ok, maybe we try a different approach. We could try just simulating the physics. We know a ton about physics! However, as before, there are a ton of tiny interactions to compute, and you need to take an incredibly small timestep. Even just a few milliseconds of protein folding is extraordinarily computationally expensive, requiring 1 trillion+ frames of simulation, for several thousand atoms, with several physical forces acting upon each one. It is also notoriously hard to parallelize these computations.
In short, none of these approaches scale particularly well.
ML Protein Prediction to the Rescue
Now that you’re wise to the game, you might ask yourself two very important questions: Can we use ML to predict protein structures? And can we get models to predict at scale? Yes, and yes!
In fact, this idea is the basis for the original AlphaFold paper, and it blew the previous attempts out of the water. At a very high level, AlphaFold used known information about proteins, including their 2D sequence, a genetic database, and a structural database of similar proteins, combined with a relatively straightforward neural net, to predict the 3D shape. After a few more tweaks in AlphaFold 2, the model was performing just about as well as traditional (read: slow and expensive) imaging methods. The paper is in the top-500 most cited papers of all time, in any scientific domain. Problem solved, case closed.
Or is it?
As with all good trilogies, AlphaFold 1 and 2 had a key flaw that necessitated a third entry. Namely, proteins aren’t the only game in town, or, perhaps, even the most important. Ligands, for example, are small molecules that interact with proteins. The majority of drugs are small molecules, and v.2 was not very accurate in predicting their structure, or how they interact with proteins. In a way, earlier models were able to deliver an exceptional salad and starter, when what we really needed was the meat and potatoes of drug discovery (i.e. ligands).
AlphaFold 3 was trained to predict other molecule types, such as ligands and complexes, and improve overall protein structure accuracy. This is really a story about simplification to achieve generalization.
Alpha Fold 3: Three Steps to Success
The architecture of AlphaFold 3 is, in many ways, more straightforward than its predecessors. It involves three simple steps:
Step 1: Insert input sequence and generate embeddings.
Step 2: Use a pairformer module to build more info (i.e. refine the representation) about the amino acid sequence.
Step 3: Using the amino acid info as input, ask a diffusion module where are the molecule’s atoms in space.
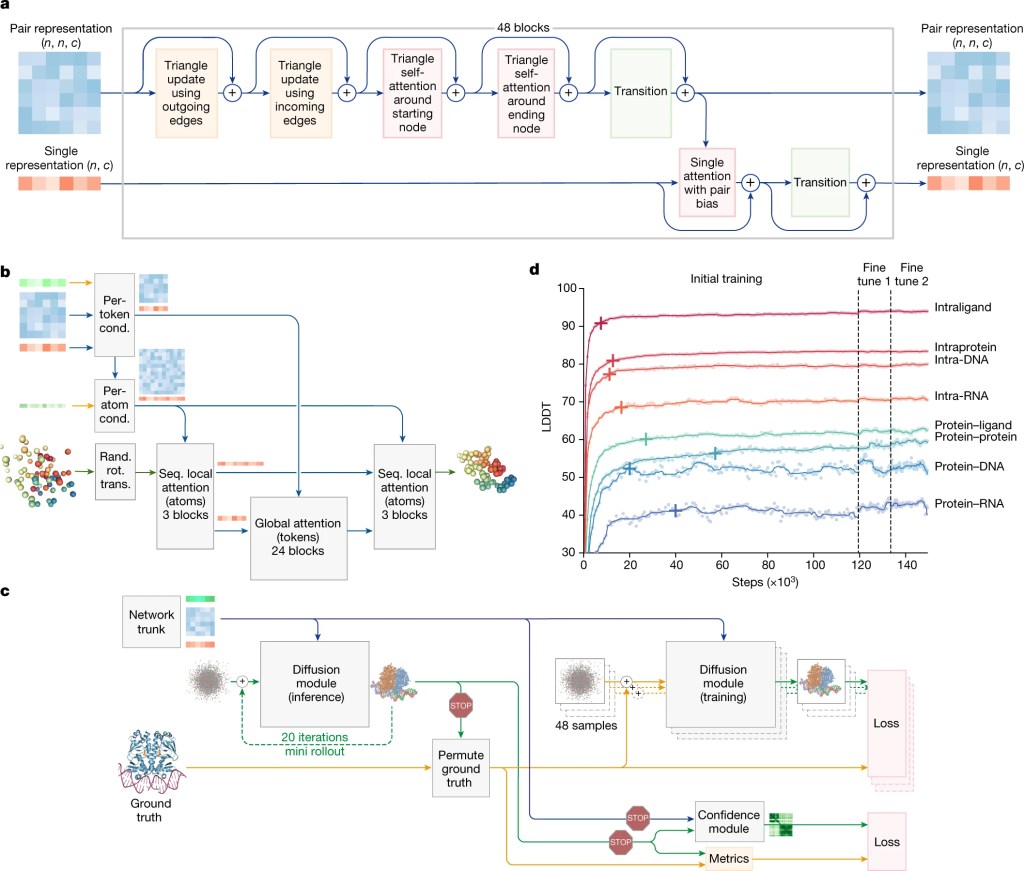
High-level architecture of Alpha Fold 3. Pairformer module (a), diffusion module (b), and training set-up (c).
At the end, the model’s output is passed through a confidence module, and voilà, we have a highly accurate and generalizable model. Let’s go through these steps one by one.
Step 1: Insert input sequence and generate embeddings
First, we start with stuff we already know about the molecule. For example, the molecule category (protein, RNA, DNA, ligand, etc.), the sequence itself as a one-hot encoding, the charge of the atom, etc. Complexes are divided up into an array of “tokens”, allowing for greater flexibility. We add to the mix the genetic database, i.e. the evolutionary history of the protein/molecule to figure out how it might fold, and template search, where we look for sequences that are similar with known structures to seed the guess.
We take this information and create two embeddings, a single embedding and a pair embedding.
Single Embedding => Straightforward vector of token features
i.e. S = [distance, residue info, … ]
Pair Embedding => Info about pairs of tokens, how they relate to each other
i.e. Z(i, j) = [bonded, same_charge, … ]
Using a single and pair representation allows for generalization. It captures what we know about an individual entity (like a small molecule) as well as how they relate with each other (as in a complex), without making a priori assumptions about how sub-components interact.
At the end of step one, we have a numerical representation of everything we know about the molecule whose structure we wish to predict, and databases (genetic and structural) about other, similar known molecules.
Step 2: Use a pairformer module to build more info (i.e. refine the representation) about the amino acid sequence
In the second phase, the embeddings are repeatedly passed through a Multiple Sequence Alignment (MSA) module, which updates the network weights using evolutionary history, and a pairwise module, which updates the pair representation of every token as it relates to every other token. The MSA module has been greatly downscaled relative to previous versions, and only has 4 blocks. In comparison, the pairwise module has 48 blocks.
The MSA algorithm is an oldie, but a goodie. You take your input protein, search for proteins that are similar to it in function, and investigate how similar their 2D sequences are. You calculate how many “jumps” (deletions, insertions, mutations) happened to go from protein A to protein B. For example, to go from S-A-D to M-A-D only requires 1 change, or mutation. To go from E-N-G-I-N-E-E-R to A-S-T-R-O-N-A-U-T requires many mutations, or, in my case, two failed NASA applications and counting. AlphaFold 2 relied heavily on MSA and attended heavily to column; in other words, if we say some amino acid A in the same position in proteins with similar functions. AlphaFold 3 cares much less. On a high level, it takes the mean down the column (i.e. what’s the most common amino acid at position N). It then sends this information into the pairwise representation, which does the heavy lifting.
This represents a shift towards generalizability, as the model relies less on seeding with evolution research (which, again, isn’t applicable for all molecules), and more on token pairs. Basically, the later is an attention network in a triangle shape. In other words, the model attends to two tokens (i, j) and some third token k. Remember that these token pairs are representations of amino acids, and how they interact with each other. As we make an update to i relative to the properties of j, it must be true that for every k, the distances in the triangle (i, j, k) do not break the triangle property. The model asks the question – how much should i pay attention to j? How much can it do so while still being a valid triangle with k? Simple geometry at the end of the day.
Taken in conjunction, this represents a new and simpler way of doing things. Instead of teaching the model, as in v.2, “a cat has 4 legs, and a cat and dog are similar in that they both have 4 legs and a tail”, just feed in pictures of cats and dogs and let the model learn structure, and how dogs and cats are similar. So, at the end of step 2, we’ve fed the embeddings into the (tiny) MSA module and (giant) pairwise module to update the weights and come up with a more refined description of the amino acids, and where they are in relation to each other. This is our “rough draft” description of the molecule.

Fig 1. Learning implicitly how a cat is like a dog, or like a human, is often a better model architecture than explicitly feeding in facts. Like, how a Maine Coon cat is huge at a length of > 1 meter, or 1.3 yards.
Step 3: Using the amino acid info as input, ask a diffusion module where are the molecule’s atoms in space.
In previous versions of Alpha Fold, step 3 was complicated. It kept track of amino-acid specific frames and angles between atoms. As you might remember from the introduction, keeping track of lots of specialized interactions based on prior knowledge doesn’t tend to scale well. In Alpha Fold 3, researchers decided to do away with the angles approach entirely, and just think about the positions of the atoms. They needed a way to take a rough description (see step 2) of a molecule, and come up with a well-defined and accurate 3D shape. They looked around and asked if there were any other ML domains that tackled this problem.
If you remember way back to 2021, when diffusion models hit it big time, everyone was like “Woah, woah, what if you had a giraffe in space, and he was reading Atlas Shrugged? And wearing a party hat!” Yeah, those diffusion models. Since we can think of atoms like real-world pixels, the problems of goofy giraffe creation and the problem of drug discovery were oddly similar. There’s no direct hard coding info about how giraffes behave, or what Atlas Shrugged is about, or whether or not a giraffe has ever read a book. However, learned embeddings from a large corpus passed through attention networks, plus a prompt about the desired output can result in a picture of said goofy giraffe. In other words, diffusion solves the problem of how to arrange a set of pixels that forms a coherent image from a start point of knowledge (the prompt), in a way that generalizes well.

Fig. 1. Aforementioned giraffe. Surprisingly terrifying. Image generated via DALL-E.
In a diffusion algorithm, you want to go from description -> pixel positions. To build the model, you take known structures (e.g. a picture of a cat) and add some noise. Next, you train an algorithm to de-noise from the noisy atoms + the correct description (“Show me a cat!”) with gradient descent. Over many iterations, this denoising model leads you to the actual pixel positions (the unscrambled cat). In broad strokes, the model learns the most likely position for each atom. With this handy dandy model, you can take pixels where positions are truly just random noise, add in the desired description, and end up with the correct position (again, the cat) within n loops. In the case of our molecules, at inference time, we sample random noise and recurrently de-noise to produce the final, highly accurate, structure. Ba-da-bing, ba-da-boom, we have our 3D structure!
This approach worked quite well. PoseBusters* is the canonical test set for 3D structure prediction. For ligands, the previous state of the art was ~50% accuracy, and it soared to nearly 80% with AlphaFold 3. Similar gains were shown with nucleic acids. Even with proteins, AlphaFold 3 beat out v.2 with p < 0.001. This is impressive considering the researchers “took away” a fair amount of domain knowledge and special cases.
* Terrible name, if you ask me
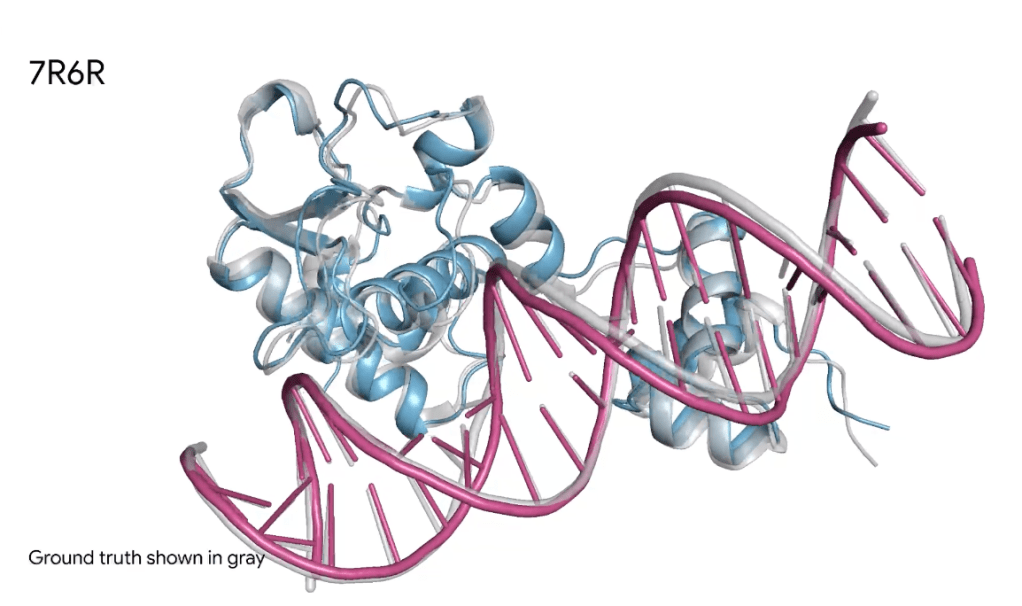
Fig. 1. Protein in blue bound to DNA double helix in pink. The blue and pink represent Alpha Fold 3 predictions, and the grey is ground truth. The prediction and ground truth are nearly identical.
The result is highly accurate 3D structure prediction across a wide variety of molecules.
The Fine Print
Now wait, you say, this feels a little too much like magic. I pop in a few details about a molecule, and out pops the correct structure, every single time? You’d be right to be skeptical. For one, diffusion is generative. Which means a few things:
1) The model produces a distribution of answers. Local structure may be well-defined, even if the model isn’t totally sure about every atom’s position. Similarly, it is somewhat sensitive to random seed.
2) It is prone to hallucination.
The second point should be familiar to anyone who has every been told by the all-knowing AI to add glue to pizza (aside: GenAI models are still terrible at interpreting humor!). To get around this, the researchers enriched AlphaFold 3 with structures predicted by AlphaFold 2. They also “taught” v.3 to represent unstructured regions as long loops instead of completely making up an answer.
Lastly, after noticing the model doesn’t respect chirality (like how you have a left hand and right hand, so do proteins), they introduced a penalty in the ranking formula for chirality violation. It still got chirality wrong 4.4% of the time.
This process of post-hoc information insertion does somewhat go against the whole generalizability claim, since it’s still relying on the older, more specific model to insert the proverbial bumpers in the bowling alley. The 3D structure won’t veer too off course, but this means of achieving success doesn’t look very cool, does it? In any case, it’s a small gripe for a model that does perform exceptionally well across a wide variety of possible molecules.
AlphaFold 3: The Simple Life
AlphaFold 3 can now predict structures of many, many molecules very accurately, not just proteins. While there are many factors in successful drug discovery beyond the scope of this blog post, predicting all manner of 3D molecular structures represents massive progress.
It generalizes by simplifying in three steps:
1) Gather info about the molecule and embed.
2) Rely heavily on pairwise transformers, less on evolutionary and expert data -> Learn patterns from data.
3) Use the prompt from step 2 and a diffusion model to predict atom locations, resulting in the 3D structure.
You can check out AlphaFold 3 for yourself here. Thanks for reading, and thanks in particular to the resources consulted when researching for this post (3Blue1Brown, Looking Glass Universe).
Catch you next time.
